BRAFV600E変異神経膠腫の分子標的治療法に対する 薬剤抵抗性機序の解明と耐性化を克服する治療標的分子の同定
2022.03.30
- プレスリリース
- 研究
BRAFV600E変異神経膠腫の分子標的治療法に対する 薬剤抵抗性機序の解明と耐性化を克服する治療標的分子の同定
横浜市立大学附属病院 脳神経外科 立石健祐 診療講師、笹目丈 医師(大学院生)、山本哲哉 主任教授らを中心とした研究グループは、患者腫瘍検体から樹立したヒト由来脳腫瘍マウスモデル(PDXモデル)を活用したトランスレーショナル研究を通じて、BRAFV600E 変異高悪性度神経膠腫の薬剤抵抗性機序を解明するとともに、耐性化を克服する標的分子と治療候補薬剤を見出しました。今回の研究成果を通じてBRAFV600E変異神経膠腫に対する臨床面での今後の展開が期待されます。
本研究は、米国がん学会誌 Clinical Cancer Researchに掲載されました。(3月28日オンラインファースト)
本研究は、米国がん学会誌 Clinical Cancer Researchに掲載されました。(3月28日オンラインファースト)
研究成果のポイント
|
研究の背景
神経膠腫*1は最も頻度の高い脳腫瘍のひとつであり、悪性度が生命予後と密接に関連する疾患であることが知られています。神経膠腫に対する診断・治療アプローチとして、外科手術による病変の摘出に始まり、術後は摘出した病変を用いて、病理組織学的診断と分子遺伝子学的診断を組み合わせた統合的な診断化を図ります。この統合診断結果に基づき、疾病の分類化を図り、術後放射線治療や化学療法の併用を検討します。このような治療アプローチに加えて、近年では遺伝子異常をターゲットにした分子標的治療が新たな治療法として期待されています。しかし、遺伝子異常を標的とした新たな治療法の開発は試みられているものの、現時点では脳腫瘍患者さんの治療成績の向上につながる画期的な治療法の確立には至っていません。
近年の大規模遺伝子解析により、人の悪性腫瘍の形成に関連する遺伝子異常が数多く見いだされています。中でも、細胞増殖に関わるタンパク質のひとつであるBRAFにおいて、BRAF遺伝子のコドン600番目の点突然変異 (BRAF:c.1799T>A p.V600E)は代表的ながん遺伝子として知られています。BRAFV600E変異が生じることで細胞内のBRAF分子の恒常的な活性化が生じ、悪性腫瘍の重要なシグナル伝達経路であるMEK-ERK (MAPK) 経路*2が過剰に活性化し、腫瘍形成が促進すると考えられています。特に重要な点としてBRAFV600E変異に対しては、BRAF阻害剤やBRAFの下流に位置するMEKを標的とした阻害剤を用いた分子標的治療が既に多くの固形がん治療に用いられており、これまでに良好な治療効果が報告されています。同様に、BRAFV600E変異は脳腫瘍にも存在することから、脳腫瘍患者さんの治療においても、BRAFV600E変異の有無に応じて治療法を個別に提唱することが期待されています。しかし、悪性度の高い神経膠腫では分子標的薬剤治療後の耐性化といった問題が指摘されており、また耐性化の原因については未だ不明な点が多く残るのが現状です。
これらの問題を解決すべく、本研究ではBRAFV600E変異神経膠腫に対する薬剤治療後の耐性化、抵抗性の機序の解明を図るとともに、抵抗性機序を克服する分子機序の同定、さらには分子機序に基づく治療法の開発を目指しました。とりわけ、臨床像の再現性の高いモデルを確立するため、分子標的の治療前後に採取したペア腫瘍検体から細胞株の樹立、およびヒト由来脳腫瘍マウスモデル (patient-derived xenograft, PDX) *3の作成を試みました。
近年の大規模遺伝子解析により、人の悪性腫瘍の形成に関連する遺伝子異常が数多く見いだされています。中でも、細胞増殖に関わるタンパク質のひとつであるBRAFにおいて、BRAF遺伝子のコドン600番目の点突然変異 (BRAF:c.1799T>A p.V600E)は代表的ながん遺伝子として知られています。BRAFV600E変異が生じることで細胞内のBRAF分子の恒常的な活性化が生じ、悪性腫瘍の重要なシグナル伝達経路であるMEK-ERK (MAPK) 経路*2が過剰に活性化し、腫瘍形成が促進すると考えられています。特に重要な点としてBRAFV600E変異に対しては、BRAF阻害剤やBRAFの下流に位置するMEKを標的とした阻害剤を用いた分子標的治療が既に多くの固形がん治療に用いられており、これまでに良好な治療効果が報告されています。同様に、BRAFV600E変異は脳腫瘍にも存在することから、脳腫瘍患者さんの治療においても、BRAFV600E変異の有無に応じて治療法を個別に提唱することが期待されています。しかし、悪性度の高い神経膠腫では分子標的薬剤治療後の耐性化といった問題が指摘されており、また耐性化の原因については未だ不明な点が多く残るのが現状です。
これらの問題を解決すべく、本研究ではBRAFV600E変異神経膠腫に対する薬剤治療後の耐性化、抵抗性の機序の解明を図るとともに、抵抗性機序を克服する分子機序の同定、さらには分子機序に基づく治療法の開発を目指しました。とりわけ、臨床像の再現性の高いモデルを確立するため、分子標的の治療前後に採取したペア腫瘍検体から細胞株の樹立、およびヒト由来脳腫瘍マウスモデル (patient-derived xenograft, PDX) *3の作成を試みました。
研究内容
BRAFV600E変異神経膠腫細胞株とマウス腫瘍モデルの樹立
研究者らのグループでは、臨床検体を用いて遺伝子解析を常時行っており、脳腫瘍の統合診断化を図っています。今回解析した217例の神経膠腫のうち、7例 (3.2%) の腫瘍からBRAFV600E変異が検出されました。この中で再発時にBRAF阻害剤 (dabrafenib)もしくはMEK阻害剤(trametinib)による分子標的治療を施行した症例を2例経験しました。1例目 (YMG62)はBRAF阻害剤とMEK阻害剤による併用治療を行いましたが、有効な治療効果は認めませんでした。2例目 (YMG89)ではBRAF阻害剤による分子標的治療を行い、当初は良好な治療効果を得たものの、経過中に耐性化が認められました。共同研究施設での症例 (NGT41)においても、2例目と同様に分子標的治療当初は良好な治療効果が認められました (Kanemaru Y et al. Acta Neuropathol Commun, 2019)。
YMG89において、治療後に耐性化をきたした検体(YMG89R)では、BRAFが関連するシグナル伝達経路であるRAS-RAF-MEK-ERK (MAPK) 経路を構成する分子をコードするNRAS, MAP2K1遺伝子変異が検出されました。しかしながら非常に低い変異率であったことからも耐性の主たる要因とは考えにくい所見でした。そこで、分子標的治療前後で採取した腫瘍組織をタンパク質レベルで比較検討したところ、MAPK経路やAKT−mTOR経路、更には上流のreceptor tyrosine kinase(RTK) 経路のリン酸化タンパク質の発現が分子標的治療後に高発現しており、再発例ではこれらのシグナル伝達経路が過剰に活性化していることが示唆されました。
次に、分子標的前後に採取した2種類の腫瘍細胞 (P; 治療前, R; 治療後)と分子標的治療後に採取したNGT41R腫瘍細胞に対して細胞培養を行ったところ、細胞株として安定化が得られました。また、腫瘍細胞を免疫不全マウス(SCID Beige)の脳内に移植したところ、ヒト由来BRAFV600E 変異神経膠腫細胞株(PDX)モデルの作成に成功しました。これらのモデルは病理学的検討、分子遺伝学的な解析を通じて患者腫瘍の遺伝型、表現型を高いレベルで再現するモデルであることを確認しました(図1)。
研究者らのグループでは、臨床検体を用いて遺伝子解析を常時行っており、脳腫瘍の統合診断化を図っています。今回解析した217例の神経膠腫のうち、7例 (3.2%) の腫瘍からBRAFV600E変異が検出されました。この中で再発時にBRAF阻害剤 (dabrafenib)もしくはMEK阻害剤(trametinib)による分子標的治療を施行した症例を2例経験しました。1例目 (YMG62)はBRAF阻害剤とMEK阻害剤による併用治療を行いましたが、有効な治療効果は認めませんでした。2例目 (YMG89)ではBRAF阻害剤による分子標的治療を行い、当初は良好な治療効果を得たものの、経過中に耐性化が認められました。共同研究施設での症例 (NGT41)においても、2例目と同様に分子標的治療当初は良好な治療効果が認められました (Kanemaru Y et al. Acta Neuropathol Commun, 2019)。
YMG89において、治療後に耐性化をきたした検体(YMG89R)では、BRAFが関連するシグナル伝達経路であるRAS-RAF-MEK-ERK (MAPK) 経路を構成する分子をコードするNRAS, MAP2K1遺伝子変異が検出されました。しかしながら非常に低い変異率であったことからも耐性の主たる要因とは考えにくい所見でした。そこで、分子標的治療前後で採取した腫瘍組織をタンパク質レベルで比較検討したところ、MAPK経路やAKT−mTOR経路、更には上流のreceptor tyrosine kinase(RTK) 経路のリン酸化タンパク質の発現が分子標的治療後に高発現しており、再発例ではこれらのシグナル伝達経路が過剰に活性化していることが示唆されました。
次に、分子標的前後に採取した2種類の腫瘍細胞 (P; 治療前, R; 治療後)と分子標的治療後に採取したNGT41R腫瘍細胞に対して細胞培養を行ったところ、細胞株として安定化が得られました。また、腫瘍細胞を免疫不全マウス(SCID Beige)の脳内に移植したところ、ヒト由来BRAFV600E 変異神経膠腫細胞株(PDX)モデルの作成に成功しました。これらのモデルは病理学的検討、分子遺伝学的な解析を通じて患者腫瘍の遺伝型、表現型を高いレベルで再現するモデルであることを確認しました(図1)。
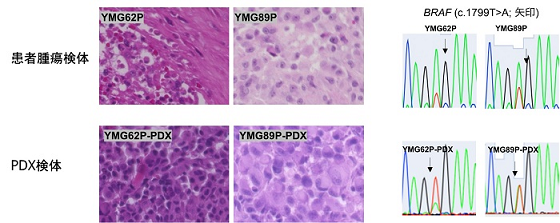
図1 BRAFV600E 変異神経膠腫検体と再現性の⾼い PDX モデル(ヘマトキシリンエオジン染⾊とBRAFV600E 変異解析)
分子標的治療後の主たる耐性化機序はフィードバック機構によるMAPK経路やAKT−mTOR経路の活性化である
樹立したBRAFV600E変異神経膠腫細胞株を用いて、細胞レベル、動物レベルで分子標的治療後の腫瘍細胞における細胞内シグナル挙動の検討を行った結果、BRAF阻害剤やMEK阻害剤に耐性化した腫瘍では、MAPK経路やAKT−mTOR経路のリン酸化タンパク質が分子標的治療後に高発現していることが明らかになりました(図2)。このことから、BRAFV600E変異神経膠腫細胞における分子標的治療の耐性機序はRTK経路、MAPK経路やAKT−mTOR経路のフィードバック機構による過剰な活性化が主たる要因であることが判明しました。
樹立したBRAFV600E変異神経膠腫細胞株を用いて、細胞レベル、動物レベルで分子標的治療後の腫瘍細胞における細胞内シグナル挙動の検討を行った結果、BRAF阻害剤やMEK阻害剤に耐性化した腫瘍では、MAPK経路やAKT−mTOR経路のリン酸化タンパク質が分子標的治療後に高発現していることが明らかになりました(図2)。このことから、BRAFV600E変異神経膠腫細胞における分子標的治療の耐性機序はRTK経路、MAPK経路やAKT−mTOR経路のフィードバック機構による過剰な活性化が主たる要因であることが判明しました。
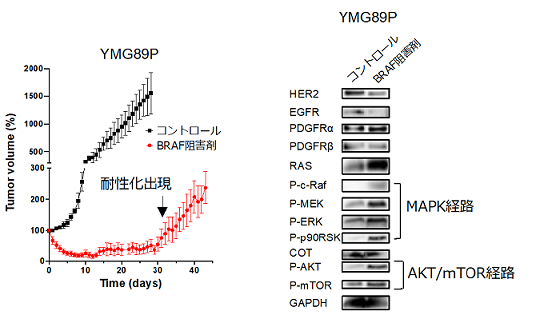
図 2 BRAFV600E 変異神経膠腫動物モデルを用いた腫瘍制御効果(左)と治療によるタンパク質発現変動(右)
HSP90はBRAFV600E変異神経膠腫に対するBRAF、MEKを標的とした分子標的治療後の耐性化を克服する標的分子である
今回の実験を通じて様々な角度から検証したところ、BRAFV600E変異神経膠腫細胞の活性を抑制するためにはMAPK経路とAKT-mTOR経路を同時にかつ強力に阻害する必要性が見いだされました。
そこで治療候補薬剤を探索するため、分子標的薬に耐性を示すYMG89R細胞に対し、分子標的薬との併用効果について1,000種類以上の化合物を用いて網羅的に検証したところ、複数のHSP90*4阻害剤がBRAF阻害剤との併用効果を示しました。このことから、HSP90阻害剤は分子標的薬との併用効果を示す薬剤である可能性が示唆されました。
そのため、複数のBRAFV600E変異神経膠腫細胞株においてBRAF阻害剤、MEK阻害剤とHSP90阻害剤 (BIIB021)との併用効果を検証した結果、単独での抗腫瘍効果は乏しく、併用治療により強い相乗効果が生まれることが判明しました。このことから、BRAF阻害剤またはMEK阻害剤にHSP90阻害剤を併用することで、MAPK経路とAKT-mTOR経路が同時かつ強力に抑制され、その結果相乗的な細胞活性抑制が生じることが明らかになりました。
今回の実験を通じて様々な角度から検証したところ、BRAFV600E変異神経膠腫細胞の活性を抑制するためにはMAPK経路とAKT-mTOR経路を同時にかつ強力に阻害する必要性が見いだされました。
そこで治療候補薬剤を探索するため、分子標的薬に耐性を示すYMG89R細胞に対し、分子標的薬との併用効果について1,000種類以上の化合物を用いて網羅的に検証したところ、複数のHSP90*4阻害剤がBRAF阻害剤との併用効果を示しました。このことから、HSP90阻害剤は分子標的薬との併用効果を示す薬剤である可能性が示唆されました。
そのため、複数のBRAFV600E変異神経膠腫細胞株においてBRAF阻害剤、MEK阻害剤とHSP90阻害剤 (BIIB021)との併用効果を検証した結果、単独での抗腫瘍効果は乏しく、併用治療により強い相乗効果が生まれることが判明しました。このことから、BRAF阻害剤またはMEK阻害剤にHSP90阻害剤を併用することで、MAPK経路とAKT-mTOR経路が同時かつ強力に抑制され、その結果相乗的な細胞活性抑制が生じることが明らかになりました。
動物実験を通じて示されたHSP90阻害剤とMEK阻害剤の相乗的な抗腫瘍効果
細胞レベルで得られた併用治療効果を動物モデルで検証するために、複数のBRAFV600E変異神経膠腫細胞株をマウス皮下に移植したPDXモデルを作成しました。その結果、細胞モデルと同様に、MEK阻害剤とHSP90阻害剤の併用療法では腫瘍が退縮後に消失し、この所見は長期間にわたり維持されました。また、マウス脳腫瘍モデルを用いて治療効果を検討したところ、皮下腫瘍モデルと同様にMEK阻害剤とHSP90阻害剤による併用療法により、MEK単剤治療と比較して更に有意な生存期間の延長効果が確認されました(図3)。
このことからBRAFV600E変異神経膠腫に対するHSP90阻害剤とMEK阻害剤による併用療法は、動物モデルにおける皮下病変及び脳病変に対して有効な治療効果を発揮することが明らかになりました。
細胞レベルで得られた併用治療効果を動物モデルで検証するために、複数のBRAFV600E変異神経膠腫細胞株をマウス皮下に移植したPDXモデルを作成しました。その結果、細胞モデルと同様に、MEK阻害剤とHSP90阻害剤の併用療法では腫瘍が退縮後に消失し、この所見は長期間にわたり維持されました。また、マウス脳腫瘍モデルを用いて治療効果を検討したところ、皮下腫瘍モデルと同様にMEK阻害剤とHSP90阻害剤による併用療法により、MEK単剤治療と比較して更に有意な生存期間の延長効果が確認されました(図3)。
このことからBRAFV600E変異神経膠腫に対するHSP90阻害剤とMEK阻害剤による併用療法は、動物モデルにおける皮下病変及び脳病変に対して有効な治療効果を発揮することが明らかになりました。
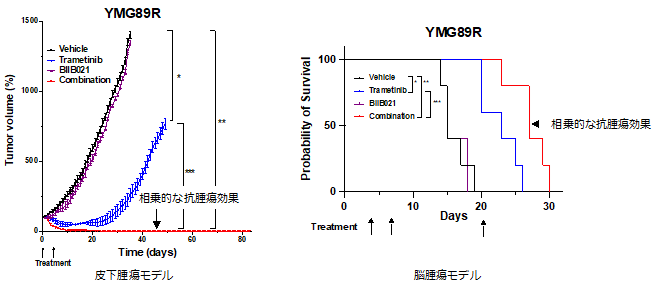
図 3 動物モデルを用いたMEK阻害剤とHSP90阻害剤併用による抗腫瘍効果の検証
今後の展開
今回の研究では、分子標的治療前後の臨床情報を活用するとともに、臨床像を広く模倣する細胞株、動物モデルを樹立し、これらを研究に活用したことで、病態の解明や治療標的分子の探索といった詳細な検討が可能になりました。今回の研究結果により、BRAFV600E変異高悪性度神経膠腫に対する分子標的治療後の主たる耐性化機序は、フィードバック機構によるMAPK経路やAKT−mTOR経路の過剰な活性化であること、またHSP90はBRAF、MEKを標的とした分子標的治療後の耐性化を克服する重要な標的分子であることが判明しました(図4)。このことから、分子標的治療による効果が限定的な症例や耐性化が見られる症例では、HSP90阻害剤の併用により、腫瘍抑制効果が得られることが期待されます。
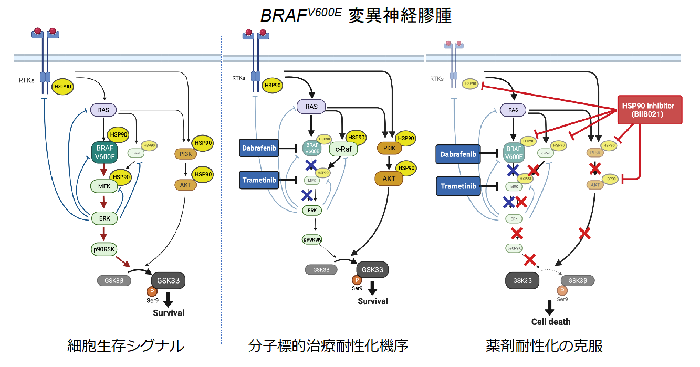
図 4 分子標的治療耐性化機序と耐性化克服のための治療アプローチ
研究費
本研究は、科学研究費補助金(19K09488、19K09476)、国立研究開発法人日本医療研究開発機構(AMED, JPcm0106502, JPck0106607)、横浜市立大学第5期戦略的研究推進事業、横浜総合医学振興財団 がん研究助成、新潟大学共同利用・共同研究(21002)等による支援を得て行われました。
論文情報
タイトル:HSP90 inhibition overcomes resistance to molecular targeted therapy in BRAFV600E mutant high-grade glioma
著者:Jo Sasame1, Naoki Ikegaya1, Masahito Kawazu2,3, Manabu Natsumeda4, Takahiro Hayashi1, Masataka Isoda1, Kaishi Satomi5, Arata Tomiyama6, Akito Oshima1, Hirokuni Honma1, Yohei Miyake1, Katsuhiro Takabayashi1, Taishi Nakamura1, Toshihide Ueno2, Yuko Matsushita6, Hiromichi Iwashita7, Yu Kanemaru4, Hidetoshi Murata1, Akihide Ryo8, Keita Terashima9, Shoji Yamanaka7, Yukihiko Fujii4, Hiroyuki Mano2, Takashi Komori10, Koichi Ichimura6, Daniel P. Cahill11, Hiroaki Wakimoto11, Tetsuya Yamamoto1, Kensuke Tateishi1*
1. 横浜市立大学大学院医学研究科 脳神経外科学
2. 国立がん研究センター細胞情報学分野
3. 千葉県がんセンター細胞治療開発研究部
4. 新潟大学脳研究所脳神経外科学分野
5. 国立がん研究センター病理診断科
6. 順天堂大学脳疾患連携分野研究講座
7. 横浜市立大学附属病院病理診断科・病理部
8. 横浜市立大学微生物学・分子生体防御学
9. 国立成育医療研究センター小児がんセンター
10. 東京都立神経病院臨床研究室
11. マサチューセッツ総合病院・ハーバード大学脳神経外科
*責任著者
掲載雑誌:Clinical Cancer Research
DOI:10.1158/1078-0432.CCR-21-3622
著者:Jo Sasame1, Naoki Ikegaya1, Masahito Kawazu2,3, Manabu Natsumeda4, Takahiro Hayashi1, Masataka Isoda1, Kaishi Satomi5, Arata Tomiyama6, Akito Oshima1, Hirokuni Honma1, Yohei Miyake1, Katsuhiro Takabayashi1, Taishi Nakamura1, Toshihide Ueno2, Yuko Matsushita6, Hiromichi Iwashita7, Yu Kanemaru4, Hidetoshi Murata1, Akihide Ryo8, Keita Terashima9, Shoji Yamanaka7, Yukihiko Fujii4, Hiroyuki Mano2, Takashi Komori10, Koichi Ichimura6, Daniel P. Cahill11, Hiroaki Wakimoto11, Tetsuya Yamamoto1, Kensuke Tateishi1*
1. 横浜市立大学大学院医学研究科 脳神経外科学
2. 国立がん研究センター細胞情報学分野
3. 千葉県がんセンター細胞治療開発研究部
4. 新潟大学脳研究所脳神経外科学分野
5. 国立がん研究センター病理診断科
6. 順天堂大学脳疾患連携分野研究講座
7. 横浜市立大学附属病院病理診断科・病理部
8. 横浜市立大学微生物学・分子生体防御学
9. 国立成育医療研究センター小児がんセンター
10. 東京都立神経病院臨床研究室
11. マサチューセッツ総合病院・ハーバード大学脳神経外科
*責任著者
掲載雑誌:Clinical Cancer Research
DOI:10.1158/1078-0432.CCR-21-3622
用語説明
*1 神経膠腫:脳実質内に発生する腫瘍であり、比較的悪性度の低い疾患から非常に悪性度の高い疾患まで多岐に渡る。
*2 MEK-ERK (MAPK) 経路:RTK-RAS-RAF-MEK-ERKなどの主要な分子を活性化することで伝達されるシグナル経路。BRAFV600E変異などの存在により過剰に活性化される。
*3 PDX:patient-derived xenograft(異種同所性腫瘍モデル): 免疫不全マウスの脳内に患者腫瘍を移植して作成した腫瘍モデル。患者腫瘍の表現型、遺伝型を広範囲に再現する。
*4 HSP90:がんの増殖・生存に関わる多くのクライアントタンパク質の機能維持に重要な役割を果たすタンパク質。EGFRなどのRTK経路、AKTやmTORなどのAKT-mTOR経路、c-RafやMEK1などMAPK経路のクライアントタンパク質を制御する
*2 MEK-ERK (MAPK) 経路:RTK-RAS-RAF-MEK-ERKなどの主要な分子を活性化することで伝達されるシグナル経路。BRAFV600E変異などの存在により過剰に活性化される。
*3 PDX:patient-derived xenograft(異種同所性腫瘍モデル): 免疫不全マウスの脳内に患者腫瘍を移植して作成した腫瘍モデル。患者腫瘍の表現型、遺伝型を広範囲に再現する。
*4 HSP90:がんの増殖・生存に関わる多くのクライアントタンパク質の機能維持に重要な役割を果たすタンパク質。EGFRなどのRTK経路、AKTやmTORなどのAKT-mTOR経路、c-RafやMEK1などMAPK経路のクライアントタンパク質を制御する
