



2024-04-25

As a part of the activities in commemoration of the 160th anniversary of Japan-German relationship, and the 10th anniversary of partnership between the cities of Yokohama and Frankfurt, Dr. Clemens von Goetze, Ambassador of Germany to Japan, was invited on December 16, 2021 to give an online lecture titled “Germany and Japan: Partners in Overcoming the Challenges of the 21st Century”.
2023-10-26
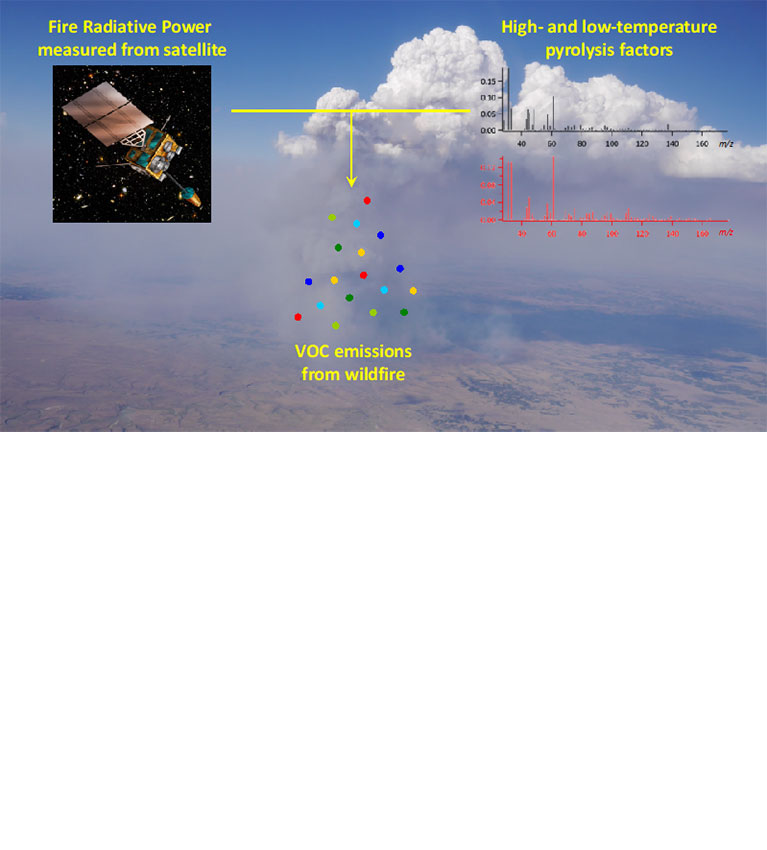
Researchers find that information on combustion temperatures can help in estimating the emission of volatile organic compounds from wildfires
2023-10-10

The 11th International Academic Consortium for Sustainable Cities (IACSC) International Symposium/General Assembly was held on November 14th, 2020, online and at Vietnam National University in Vietnam.
2023-08-29

As a part of the activities in commemoration of the 160th anniversary of Japan-German relationship, and the 10th anniversary of partnership between the cities of Yokohama and Frankfurt, Dr. Clemens von Goetze, Ambassador of Germany to Japan, was invited on December 16, 2021 to give an online lecture titled “Germany and Japan: Partners in Overcoming the Challenges of the 21st Century”.


Hospitals & Research Centers





Study at YCU
YCU's 5 undergraduate schools and 6 graduate schools, coupled with the low student-faculty ratio ensures that all our students get a quality education in a comfortable environment.
Areas of study >
Undergraduate admissions >
Graduate admissions >
International exchange >
Work at YCU
Every employee has the opportunity to make an impact supporting YCU's mission of teaching and research. See all the current vacancies available at Yokohama City University.
Giving to YCU
Thank you for your interest in making a gift to Yokohama City University. Below you can find out more about the various ways by which you can make your gift.
Give to YCU >